All Projects
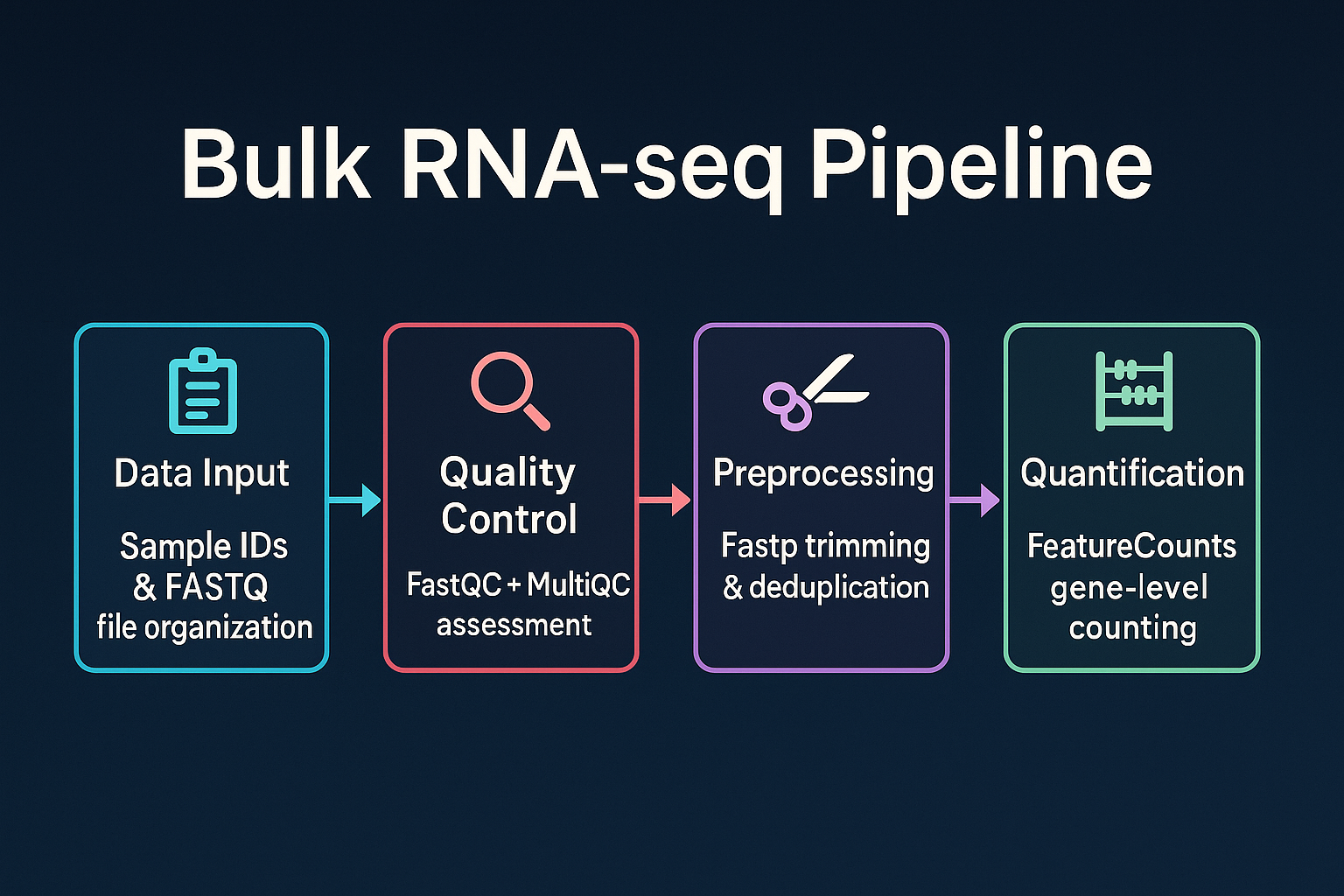
A Snakemake-based pipeline for preprocessing, quality control, alignment, quantification, and transformation of bulk RNA-seq data. Designed for flexibility and reproducibility, it supports both single-end and paired-end reads and produces normalized count matrices ready for downstream analysis.
Design & Planning
Problem framing
Lab members needed a reproducible, low-friction pipeline for bulk RNA-seq that could run on HPC clusters without manual step management. Ad-hoc scripts were fragile across different user environments and made it difficult to audit exactly which steps ran on which samples.
Key design decisions
- Snakemake for DAG-based reproducibility — automatically tracks dependencies between steps and only re-runs affected steps when inputs change, making reruns after partial failures cheap.
- Modular rules for swappable aligners — separating each tool into its own Snakemake rule allows the aligner or quantifier to be swapped with minimal changes to the workflow.
Architecture overview
[TODO: fill in architecture and challenges]
Challenges & what I’d do differently
[TODO: fill in architecture and challenges]
Features
- Download and organize raw FASTQ files
- Quality control with FastQC and MultiQC
- Adapter trimming and deduplication using Fastp
- Alignment using HISAT2
- Gene-level quantification with FeatureCounts
- Aggregation and transformation of expression data (TPM, log2TPM, VST)
- Configurable support for single- or paired-end reads
Requirements
- Snakemake
- Python >= 3.8
- External tools:
fastqc,fastp,multiqc,hisat2,samtools,featureCounts(Subread) - Python libraries:
pandas,numpy(used inaggregate_counts.py)
Directory Structure
.
├── config.yaml # Set sample IDs, paths, paired-end flag, etc.
├── Snakefile # Main Snakemake workflow
├── aggregate_counts.py # Aggregates counts and generates TPM, log2TPM, VST matrices
├── input/
│ └── sample_ids.txt # List of SRR IDs or sample names
├── raw_data/ # Input FASTQ files
├── trimmed_data/ # Output from Fastp
├── QC/
│ ├── raw/ # FastQC + MultiQC for raw data
│ └── trimmed/ # FastQC + MultiQC for trimmed data
├── alignment/ # Aligned BAM files (sorted)
├── counts/ # Count matrices (raw, TPM, log2TPM, VST)
└── ref_genomes/
└── hg38/ # Contains HISAT2 index and GTF file